拉曼光谱原理分析,拉曼光谱分析仪原理


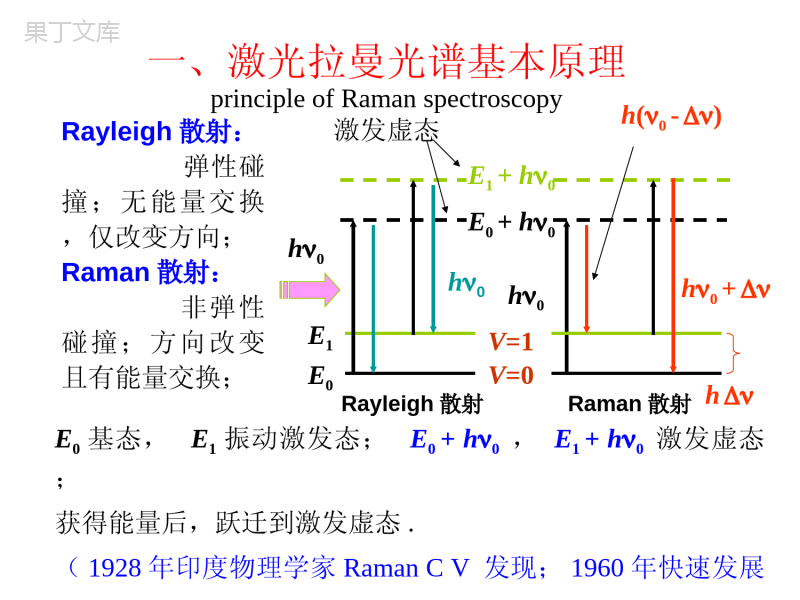
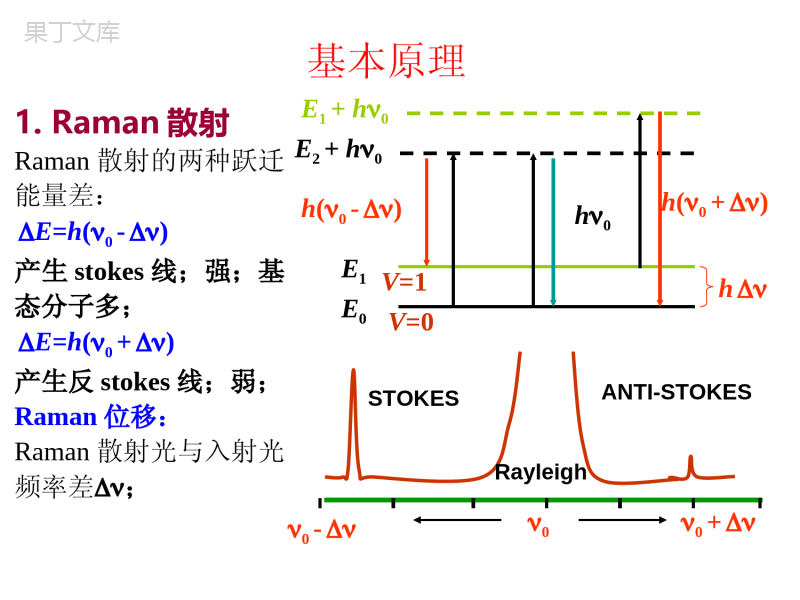

一、拉曼光谱基本原理principleofRamanspectroscopy二、拉曼光谱的应用applicationsofRamanspectroscopy三、激光拉曼光谱仪laserRamanspectroscopy第五节激光拉曼光谱分析法laserRamanspectroscopy一、激光拉曼光谱基本原理principleofRamanspectroscopyRayleigh散射:弹性碰撞;无能量交换,仅改变方向;Raman散射:非弹性碰撞;方向改变且有能量交换;Rayleigh散射Raman散射E0基态,E1振动激发态;E0+h0,E1+h0激发虚态;获得能量后,跃迁到激发虚态.(1928年印度物理学家RamanCV发现;1960年快速发展hE0E1V=1V=0h0h0h0h0+E1+h0E0+h0h(0-)激发虚态基本原理1.Raman散射Raman散射的两种跃迁能量差:E=h(0-)产生stokes线;强;基态分子多;E=h(0+)产生反stokes线;弱;Raman位移:Raman散射光与入射光频率差;ANTI-STOKES0-RayleighSTOKES0+0h(0+)E0E1V=1V=0E1+h0E2+h0hh0h(0-)2.Raman位移对不同物质:不同;对同一物质:与入射光频率无关;表征分子振-转能级的特征物理量;定性与结构分析的依据;Raman散射的产生:光电场E中,分子产生诱导偶极距=E分子极化率;3.红外活性和拉曼活性振动①红外活性振动ⅰ永久偶极矩;极性基团;ⅱ瞬间偶极矩;非对称分子;红外活性振动—伴有偶极矩变化的振动可以产生红外吸收谱带.②拉曼活性振动诱导偶极矩=E非极性基团,对称分子;拉曼活性振动—伴随有极化率变化的振动。对称分子:对称振动→拉曼活性。不对称振动→红外活性Eeer4.红外与拉曼谱图对比红外光谱:基团;拉曼光谱:分子骨架测定;红外与拉曼谱图对比对称中心分子CO2,CS2等,选律不相容。无对称中心分子(例如SO2等),三种振动既是红外活性振动,又是拉曼活性振动。5.选律SCSSCSSCS1234拉曼活性红外活性红外活性振动自由度:3N-4=4拉曼光谱—源于极化率变化红外光谱—源于偶极矩变化6.拉曼光谱与红外光谱分析方法比较拉曼光谱红外光谱光谱范围40-4000Cm-1光谱范围400-4000Cm-1水可作为溶剂水不能作为溶剂样品可盛于玻璃瓶,毛细管等容器中直接测定不能用玻璃容器测定固体样品可直接测定需要研磨制成KBR压片二、拉曼光谱的应用applicationsofRamanspectroscopy由拉曼光谱可以获得有机化合物的各种结构信息:2)红外光谱中,由CN,C=S,S-H伸缩振动产生的谱带一般较弱或强度可变,而在拉曼光谱中则是强谱带。3)环状化合物的对称呼吸振动常常是最强的拉曼谱带。1)同种分子的非极性键S-S,C=C,N=N,CC产生强拉曼谱带,随单键双键三键谱带强度增加。4)在拉曼光谱中,X=Y=Z,C=N=C,O=C=O-这类键的对称伸缩振动是强谱带,反这类键的对称伸缩振动是弱谱带。红外光谱与此相反。5)C-C伸缩振动在拉曼光谱中是强谱带。6)醇和烷烃的拉曼光谱是相似的:I.C-O键与C-C键的力常数或键的强度没有很大差别。II.羟基和甲基的质量仅相差2单位。III.与C-H和N-H谱带比较,O-H拉曼谱带较弱。2941,2927cm-1ASCH22854cm-1SCH21029cm-1(C-C)803cm-1环呼吸1444,1267cm-1CH23060cm-1r-H)1600,1587cm-1c=c)苯环1000cm-1环呼吸787cm-1环变形1039,1022cm-1单取代三、激光Raman光谱仪laserRamanspectroscopy激光光源:He-Ne激光器,波长632.8nm;Ar激光器,波长514.5nm,488.0nm;散射强度1/4单色器:光栅,多单色器;检测器:光电倍增管,光子计数器;傅立叶变换-拉曼光谱仪FT-Ramanspectroscopy光源:Nd-YAG钇铝石榴石激光器(1.064m);检测器:高灵敏度的铟镓砷探头;特点:(1)避免了荧光干扰;(2)精度高;(3)消除了瑞利谱线;(4)测量速度快。•5.6拉曼光谱•1928年,印度物理学家C.V.Raman发现光通过透明溶液时,有一部分光被散射,其频率与入射光不同,为,频率位移与发生散射的分子结构有关。这种散射称为拉曼散射,频率位移称为拉曼位移。由红外光谱及拉曼光谱可以获得分子结构的直接信息,仪器分辨率高。采用显微测定等手段可以进行非破坏、原位测定以及时间分辨测定等。•5.6.1拉曼光谱简介从图中可见,拉曼光谱的横坐标为拉曼位移,以波数表示。,其中和分别为Stokes位移和入射光波数。纵坐标为拉曼光强。由于拉曼位移与激发光无关,一般仅用Stokes位移部分。对发荧光的分子,有时用反Stokes位移。•拉曼光谱的特点:波长位移在中红外区。有红外及拉曼活性的分子,其红外光谱和拉曼光谱近似。可使用各种溶剂,尤其是能测定水溶液,样品处理简单。低波数段测定容易(如金属与氧、氮结合键的振动nM-O,nM-N等)。而红外光谱的远红外区不适用于水溶液,选择窗口材料、检测器困难。由Stokes、反Stokes线的强度比可以测定样品体系的温度。显微拉曼的空间分辨率很高,为1mm。时间分辨测定可以跟踪10-12s量级的动态反应过程。利用共振拉曼、表面增强拉曼可以提高测定灵敏度。其不足之处在于,激光光源可能破坏样品;荧光性样品测定一般不适用,需改用近红外激光激发等等。•5.6.2拉曼及瑞利散射机理•瑞利和拉曼散射的产生•测定拉曼散射光谱时,一般选择激发光的能量大于振动能级的能量但低于电子能级间的能量差,且远离分析物的紫外-可见吸收峰。当激发光与样品分子作用时,样品分子即被激发至能量较高的虚态(图中用虚线表示)。左边的一组线代表分子与光作用后的能量变化,粗线出现的几率大,细线表示出现的几率小,因为室温下大多数分子处于基态的最低振动能级。中间一组线代表瑞利(Rayleigh)散射,光子与分子间发生弹性碰撞,碰撞时只是方向发生改变而未发生能量交换。右边一组线代表拉曼散射,光子与分子碰撞后发生了能量交换,光子将一部分能量传递给样品分子或从样品分子获得一部分能量,因而改变了光的频率。能量变化所引起的散射光频率变化称为拉曼位移。由于室温下基态的最低振动能级的分子数目最多,与光子作用后返回同一振动能级的分子也最多,所以上述散射出现的几率大小顺序为:瑞利散射>Stokes线>反Stokes线。随温度升高,反Stokes线的强度增加。•拉曼活性入射光可以看成是互相垂直的电场和磁场在空间的传播。其电场强度E可用下述交变电场描述:E=E0Cos(2pn0t)(5.47)其中,E0为交变电场波的振幅,n0为激发光频率。样品分子键上的电子云与入射光电场作用时会诱导出电偶极矩P:P=aE=aE0Cos(2pn0t)(5.48)a为键的极化度。只有当键的极化度是成键原子间距离的函数,即分子振动产生的原子间距离的改变引起分子极化度变化时,才产生拉曼散射,分子才是拉曼活性的:•(5.49)a0为分子中键处于平衡位置时的极化度,req为分子中键处于平衡位置时的核间距,r为分子中键处于任意位置时的核间距。若核间距改变时产生的振动频率为n,与平衡位置比较的最大核间距为rm,则:r-req=rmcos2pnt(5.50)代入(5.49)式:(5.51)(5.52)(5.52)式第一项对应样品的瑞利散射,其频率为n0;第二项对应样品的拉曼散射,产生反Stokes位移,频率为反Stokes频率n0+n;第三项对应样品的拉曼散射,产生Stokes位移,频率为Stokes频率n0-n。(5.52)式表明,要产生拉曼散射,分子的极化度必须是核间距的函数,即da/dr0时才会观察到拉曼线,只有振动时极化度发生变化的分子才是拉曼活性的。返回页首••5.6.3拉曼光谱参数•频率即拉曼位移,一般用Stokes位移表示。是结构鉴定的重要依据。强度I拉曼散射强度(5.53)式中I0为光源强度;K为常数。当样品分子不产生吸收时,I与激发波长的4次方成反比。因此选择较短波长的激光时灵敏度高。拉曼光强与样品分子浓度成正比。•去偏振度r(depolarization)r对确定分子的对称性很有用。•拉曼光谱的入射光为激光,激光是偏振光。设入射激光沿xz平面向O点传播,O处放样品。激光与样品分子作用时可散射不同方向的偏振光,若在检测器与样品之间放一偏振器,便可分别检测与激光方向平行的平行散射光I//(yz平面)和与激光方向垂直的垂直散射光I(xy平面)。定义去偏振度(5.54)去偏振度与分子的极化度有关。如分子的极化度中各向同性部分为,各向异性部分为,则:(5.55)返回页首••对球形对称振动,,因此去偏振度r为零。即r值越小,分子的对称性越高。若分子是各向异性的,则,r=3/4。即非全对称振动的r=03/4(0.75)。因此通过测定拉曼谱线的去偏振度,可以确定分子的对称性。如前CCl4的拉曼光谱,459cm-1是由四个氯原子同时移开或移近碳原子所产生的对称伸缩振动引起,r=0.0005,去极化度很小。459cm-1线称为极化线。而218cm-1、314cm-1源于非对称振动,r=0.75。如图5.29,结晶紫据文献报导有醌式(a)和离子型(b)两种结构。在(a)式中三个苯环处于同一平面。(b)式中三个苯环因位阻关系不处在同一平面,彼此稍许错开,形成类似螺旋桨状。测定结晶紫水溶液(5x10-4M)的拉曼谱,214cm-1(结晶紫分子中心碳原子的呼吸振动)的r值接近零,可见分子的对称性很高,说明在该实验条件下结晶紫分子为离子型结构。•红外及拉曼光谱法的比较红外及拉曼光谱法的相同点在于,对于一个给定的化学键,其红外吸收频率与拉曼位移相等,均代表第一振动能级的能量。因此,对某一给定的化合物,某些峰的红外吸收波数与拉曼位移完全相同,红外吸收波数与拉曼位移均在红外光区,两者都反映分子的结构信息。•从图中可以看出,同一物质,有些峰的红外吸收与拉曼散射完全对应,但也有许多峰有拉曼散射却无红外吸收,或有红外吸收却无拉曼散射。因此,红外光谱与拉曼光谱互补,可用于有机化合物的结构鉴定。红外光谱的入射光及检测光均是红外光,而拉曼光谱的入射光大多数是可见光,散射光也是可见光。红外光谱测定的是光的吸收,横坐标用波数或波长表示,而拉曼光谱测定的是光的散射,横坐标是拉曼位移。两者的产生机理不同。红外吸收是由于振动引起分子偶极矩或电荷分布变化产生的。拉曼散射是由于键上电子云分布产生瞬间变形引起暂时极化,产生诱导偶极,当返回基态时发生的散射。散射的同时电子云也恢复原态。例如同核双原子分子NN,Cl-Cl,H-H等无红外活性却有拉曼活性是由于这些分子平衡态或伸缩振动引起核间距变化但无偶极矩改变,对振动频率(红外光)不产生吸收。但两原子间键的极化度在伸缩振动时会产生周期性变化:核间距最远时极化度最大,最近时极化度最小。由此产生拉曼位移。二氧化碳分子的对称伸缩振动(OCO)无红外活性,但可以产生周期性极化度的改变(距离近时电子云变形小,距离远时电子云变形大),因此有拉曼活性。而非对称伸缩振动(OCO)有红外活性无拉曼活性。此时一个键的核间距减小,一个键的核间距增大(一个键的极化度小,一个键的极化度大),总的结果是无拉曼活性。图5.31直线型分子的振动举例-CO2分子振动过程中的极化度以及偶极矩变化返回页首•5.6.4拉曼光谱仪•拉曼光谱仪的光源为激光光源。由于拉曼散射很弱,因此要求光源强度大,一般用激光光源。有可见及红外激光光源等。如具有308nm,351nm发射线的紫外激光器;Ar+激光器一般在488.0nm,514.5nm等可见区发光;而Nd:YaG激光器则在1064nm的近红外区使用。色散型拉曼光谱仪有多个单色器(doubleortriplemonochrometersystem)。由于测定的拉曼位移较小,因此仪器需要较高的单色性。在傅立叶变换拉曼光谱仪中,以迈克尔逊干涉仪代替色散元件,光源利用率高,可采用红外激光,用以避免分析物或杂质的荧光干扰。拉曼光谱仪的检测器为光电倍增管、多探测器(如CCD:ChargeCoupledDevice)等。••微区分析装置的应用。微区分析装置是拉曼光谱仪的一个附件,由光学显微镜、电子摄像管、显象荧光屏、照相机等组成。可以将局部样品的放大图显示在荧光屏上,用照相机拍摄样品的显微图象。如人眼球晶体中白内障病变部位的观测等。5.6.5拉曼光谱的应用•用通常的拉曼光谱可以进行半导体、陶瓷等无机材料的分析。如剩余应力分析、晶体结构解析等。拉曼光谱还是合成高分子、生物大分子分析的重要手段。如分子取向、蛋白质的巯基、卟啉环等的分析。直链CH2碳原子的折叠振动频率可由下式确定:n=2400/Nc(cm-1)。Nc为碳原从图中可以看出,不同的碳材料其拉曼光谱不同,因此可以彼此区分。•共振拉曼RRS(ResonanceRamanScattering)以分析物的紫外-可见吸收光谱峰的邻近处作为激发波长。样品分子吸光后跃迁至高电子能级并立即回到基态的某一振动能级,产生共振拉曼散射。该过程很短,约为10-14秒。而荧光发射是分子吸光后先发生振动松弛,回到第一电子激发态的第一振动能级,返回基态时的发光。荧光寿命一般为10-6-10-8秒。共振拉曼强度比普通的拉曼光谱法强度可提高102-106倍,检测限可达10-8摩尔/升,而一般的拉曼光谱法只能用于测定0.1摩尔/升以上浓度的样品。因此RRS法用于高灵敏度测定以及状态解析等,如低浓度生物大分子的水溶液测定。共振拉曼的主要不足是荧光干扰。例如用共振拉曼确定血红蛋白和细胞色素c中Fe的氧化态和Fe原子的自旋状况。此时共振拉曼仅取决于四个吡咯环的振动方式,与蛋白质有关的其它拉曼峰并不增强,在极低浓度时并不干扰。•表面增强拉曼SERS(Surface-EnhancedRamanScattering)表面增强拉曼是用通常的拉曼光谱法测定吸附在胶质金属颗粒如银、金或铜表面的样品,或吸附在这些金属片的粗糙表面上的样品。尽管原因尚不明朗,人们发现被吸附的样品其拉曼光谱的强度可提高103-106倍。如果将表面增强拉曼与共振拉曼结合,光谱强度的净增加几乎是两种方法增强的和。检测限可低至10-9-10-12摩尔/升。表面增强拉曼主要用于吸附物种的状态解析等。
提供拉曼光谱原理分析,拉曼光谱分析仪原理会员下载,编号:1701027024,格式为 xlsx,文件大小为39页,请使用软件:wps,office Excel 进行编辑,PPT模板中文字,图片,动画效果均可修改,PPT模板下载后图片无水印,更多精品PPT素材下载尽在某某PPT网。所有作品均是用户自行上传分享并拥有版权或使用权,仅供网友学习交流,未经上传用户书面授权,请勿作他用。若您的权利被侵害,请联系963098962@qq.com进行删除处理。

 下载
下载 下载
下载 下载
下载 下载
下载 下载
下载 下载
下载 下载
下载 下载
下载 下载
下载 下载
下载 下载
下载 下载
下载